جزوه تایپ شده بیوانفورماتیک
دکتر مطهره محسن پور دانشگاه پیام نور بابک باباعباسی علمی کاربردی محمد نجفی و اباذر روستا زاده میانده آزاد کاردانی کارشناسی ارشد جینمیشل
[14] [15] [16][17] [19][20]
() () [21] شناسی محاسباتی نامیده می شود . زیرشاخه های مهم در بیوانفورماتیک و زیست شناسی محاسباتی عبارتند از:
توسعه و اجرای برنامه های کامپیوتری که امکان دسترسی کارآمد، مدیریت و استفاده از انواع مختلف اطلاعات را فراهم می کند.
توسعه الگوریتمهای جدید (فرمولهای ریاضی) و معیارهای آماری که روابط بین اعضای مجموعه دادههای بزرگ را ارزیابی میکند. به عنوان مثال، روشهایی برای تعیین مکان یک ژن در یک توالی، پیشبینی ساختار و/یا عملکرد پروتئین، و خوشهبندی توالیهای پروتئین در خانوادههایی از توالیهای مرتبط وجود دارد.
هدف اولیه بیوانفورماتیک افزایش درک فرآیندهای بیولوژیکی است. با این حال، آنچه آن را از سایر رویکردها متمایز می کند، تمرکز آن بر توسعه و بکارگیری تکنیک های محاسباتی فشرده برای دستیابی به این هدف است. مثالها عبارتند از: تشخیص الگو ، دادهکاوی ، الگوریتمهای یادگیری ماشین ، و تجسم . تلاشهای تحقیقاتی عمده در این زمینه شامل همترازی جزوه بیوانفورماتیک، یافتن ژن ، مونتاژ ژنوم ، طراحی دارو ، کشف دارو ، تراز ساختار پروتئین ، پیشبینی ساختار پروتئین ، پیشبینیبیان ژن و فعل و انفعالات پروتئین-پروتئین ، مطالعات ارتباط ژنومی ، مدلسازی تکامل و تقسیم سلولی/میتوز.
بیوانفورماتیک در حال حاضر مستلزم ایجاد و پیشرفت پایگاههای داده، الگوریتمها، تکنیکهای محاسباتی و آماری و تئوری برای حل مشکلات رسمی و عملی ناشی از مدیریت و تجزیه و تحلیل دادههای بیولوژیکی است.
در طول چند دهه گذشته، پیشرفت های سریع در فناوری های تحقیقاتی ژنومی و دیگر مولکولی و تحولات در فناوری اطلاعات با هم ترکیب شده اند تا حجم عظیمی از اطلاعات مربوط به زیست شناسی مولکولی را تولید کنند. بیوانفورماتیک نامی است که به این رویکردهای ریاضی و محاسباتی داده شده است که برای درک درستی از فرآیندهای بیولوژیکی استفاده می شود.
بیوانفورماتیک
فعالیت های رایج در بیوانفورماتیک شامل نقشه برداری و تجزیه و تحلیل توالی های DNA و پروتئین، تراز کردن توالی DNA و پروتئین برای مقایسه آنها، و ایجاد و مشاهده مدل های سه بعدی از ساختارهای پروتئینی است.
ارتباط با سایر زمینه ها
بیوانفورماتیک یک رشته علمی است که شبیه محاسبات بیولوژیکی اما متمایز از آن است، در حالی که اغلب مترادف زیست شناسی محاسباتی در نظر گرفته می شود . محاسبات بیولوژیکی از مهندسی زیستی و زیست شناسی برای ساخت رایانه های بیولوژیکی استفاده می کند ، در حالی که بیوانفورماتیک از محاسبات برای درک بهتر زیست شناسی استفاده می کند. بیوانفورماتیک و زیست شناسی محاسباتی شامل تجزیه و تحلیل داده های بیولوژیکی، به ویژه DNA، RNA، و توالی پروتئین است. زمینه بیوانفورماتیک رشد انفجاری را از اواسط دهه 1990 تجربه کرد که عمدتاً توسط پروژه ژنوم انسانی و پیشرفت های سریع در فناوری توالی یابی DNA انجام شد.
تجزیه و تحلیل داده های بیولوژیکی برای تولید اطلاعات معنی دار شامل نوشتن و اجرای برنامه های نرم افزاری است که از الگوریتم های نظریه گراف ، هوش مصنوعی ، محاسبات نرم ، داده کاوی ، پردازش تصویر و شبیه سازی کامپیوتری استفاده می کنند. الگوریتم ها به نوبه خود به مبانی نظری مانند ریاضیات گسسته ، نظریه کنترل ، نظریه سیستم ، نظریه اطلاعات و آمار بستگی دارند .
تحلیل توالی
مقالههای اصلی: همترازی توالی ، پایگاهداده توالی ، و تحلیل توالی بدون تراز
از زمانی که فاژ Φ-X174 در سال 1977 توالی یابی شد، [22] توالی DNA هزاران جاندار رمزگشایی و در پایگاه های داده ذخیره شده است. این اطلاعات توالی برای تعیین ژنهایی که پروتئینها ، ژنهای RNA، توالیهای تنظیمی، موتیفهای ساختاری و توالیهای تکراری را کد میکنند، تجزیه و تحلیل میشود. مقایسه ژنها در یک گونه یا بین گونههای مختلف میتواند شباهتهای بین عملکرد پروتئین یا روابط بین گونهها را نشان دهد (استفاده از سیستماتیک مولکولی برای ساخت درختان فیلوژنتیک ). با افزایش حجم داده ها، مدت ها پیش تجزیه و تحلیل دستی توالی های DNA غیرعملی شد. برنامه های کامپیوتریاز جمله BLAST به طور معمول برای جستجوی توالی ها استفاده می شود – از سال 2008، از بیش از 260000 موجود زنده، حاوی بیش از 190 میلیارد نوکلئوتید . [23]
جزوه بیوانفورماتیک دانلود رایگان خلاصه کتاب پی دی اف
تصویر: 450 پیکسل مراحل تجزیه و تحلیل توالی
توالی یابی DNA
مقاله اصلی: توالی یابی DNA
قبل از تجزیه و تحلیل توالی ها باید از بانک ذخیره سازی داده ها به عنوان مثال Genbank به دست آیند. تعیین توالی DNA هنوز یک مشکل غیر ضروری است زیرا داده های خام ممکن است نویز داشته باشند یا تحت تأثیر سیگنال های ضعیف قرار گیرند. الگوریتمهایی برای فراخوانی پایه برای رویکردهای تجربی مختلف برای توالییابی DNA توسعه یافتهاند.
مونتاژ سکانس
مقاله اصلی: مونتاژ توالی
اکثر تکنیکهای توالییابی DNA قطعات کوتاهی از توالی را تولید میکنند که باید برای به دست آوردن توالیهای کامل ژن یا ژنوم جمع شوند. به اصطلاح تکنیک توالی تفنگ شاتگان (که برای مثال توسط موسسه تحقیقات ژنومی (TIGR) برای تعیین توالی اولین ژنوم باکتری، هموفیلوس آنفولانزا استفاده شد ) [24]توالی هزاران قطعه کوچک DNA را تولید می کند (از 35 تا 900 نوکلئوتید طول دارد، بسته به فناوری توالی یابی). انتهای این قطعات با هم همپوشانی دارند و هنگامی که توسط یک برنامه مونتاژ ژنوم به درستی در یک راستا قرار گیرند، می توان برای بازسازی ژنوم کامل استفاده کرد. توالییابی تفنگ ساچمهای دادههای توالی را به سرعت به دست میآورد، اما کار مونتاژ قطعات برای ژنومهای جزوه بیوانفورماتیک میتواند بسیار پیچیده باشد. برای ژنومی به بزرگی ژنوم انسان ، ممکن است چند روز از زمان CPU روی رایانههای با حافظه بزرگ و چند پردازندهای برای جمعآوری قطعات طول بکشد و مجموعه حاصل معمولاً حاوی شکافهای متعددی است که باید بعداً پر شوند. توالی یابی تفنگ ساچمه ای روش انتخابی برای تقریباً همه ژنوم هایی است که امروزه توالی یابی شده اند [ چه زمانی؟ ]و الگوریتم های مونتاژ ژنوم یک حوزه حیاتی از تحقیقات بیوانفورماتیک هستند.
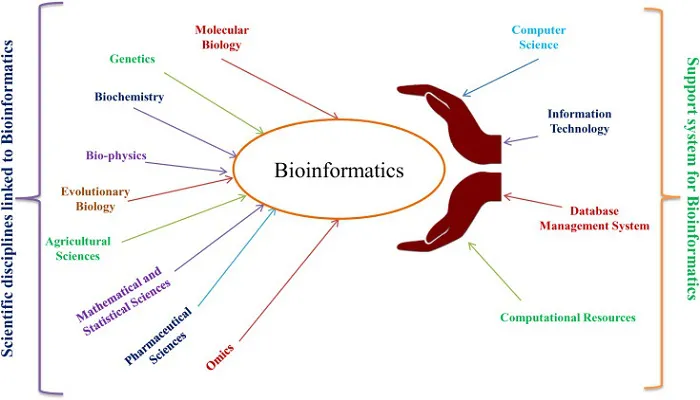
خلاصه کتاب رایگان بیوانفورماتیک
همچنین ببینید: تجزیه و تحلیل توالی ، کاوی توالی ، ابزار پروفایل سازی توالی ، و موتیف توالی
حاشیه نویسی ژنوم
مقاله اصلی: پیشبینی ژن
در زمینه ژنومیک ، حاشیه نویسی فرآیند علامت گذاری ژن ها و سایر ویژگی های بیولوژیکی در یک توالی DNA است. این فرآیند باید خودکار شود، زیرا بیشتر ژنومها برای حاشیهنویسی با دست بسیار بزرگ هستند، نه اینکه میل به حاشیهنویسی تا آنجایی که ممکن است ژنومها وجود دارد، زیرا سرعت توالیبندی دیگر باعث ایجاد گلوگاه نشده است. حاشیه نویسی با این واقعیت امکان پذیر است که ژن ها دارای مناطق شروع و توقف قابل تشخیص هستند، اگرچه توالی دقیق یافت شده در این مناطق می تواند بین ژن ها متفاوت باشد.
حاشیه نویسی ژنوم را می توان به سه سطح طبقه بندی کرد: سطوح نوکلئوتید، پروتئین و فرآیند.
یافتن ژن یکی از جنبه های اصلی حاشیه نویسی در سطح نوکلئوتید است. برای ژنومهای پیچیده، موفقترین روشها از ترکیبی از پیشبینی ژن ab initio و مقایسه توالی با پایگاههای اطلاعاتی توالی بیان شده و سایر ارگانیسمها استفاده میکنند. حاشیه نویسی در سطح نوکلئوتید همچنین امکان ادغام توالی ژنوم با سایر نقشه های ژنتیکی و فیزیکی ژنوم را فراهم می کند.
هدف اصلی حاشیه نویسی در سطح پروتئین، اختصاص عملکرد به محصولات ژنوم است. پایگاه های داده توالی های پروتئینی و حوزه ها و موتیف های عملکردی منابع قدرتمندی برای این نوع حاشیه نویسی هستند. با این وجود، نیمی از پروتئینهای پیشبینیشده در یک توالی ژنوم جدید، هیچ عملکرد آشکاری ندارند.
درک عملکرد ژن ها و محصولات آنها در زمینه فیزیولوژی جزوه ایمنی انبار و ارگانیسمی هدف حاشیه نویسی در سطح فرآیند است. یکی از موانع این سطح از حاشیه نویسی، ناهماهنگی اصطلاحات مورد استفاده در سیستم های مدل مختلف است. کنسرسیوم هستی شناسی ژن به حل این مشکل کمک می کند. [25]
اولین توصیف از یک سیستم جزوه بیوانفورماتیک حاشیه نویسی ژنوم در سال 1995 [24] توسط تیمی در موسسه تحقیقات ژنومی منتشر شد که اولین توالی یابی و تجزیه و تحلیل کامل ژنوم یک موجود زنده آزاد، باکتری هموفیلوس آنفولانزا را انجام داد. [24] Owen White یک سیستم نرم افزاری برای شناسایی ژن های کد کننده همه پروتئین ها، RNA های انتقالی، RNA های ریبوزومی (و سایر مکان ها) و انجام وظایف اولیه را طراحی و ساخت. اکثر سیستم های حاشیه نویسی ژنوم فعلی به طور مشابه کار می کنند، اما برنامه های موجود برای تجزیه و تحلیل DNA ژنومی، مانند برنامه GeneMark برای یافتن ژن های کد کننده پروتئین در هموفیلوس آنفولانزا آموزش دیده و استفاده می شود.، دائما در حال تغییر و بهبود هستند.
به دنبال اهدافی که پروژه ژنوم انسانی پس از بسته شدن آن در سال 2003 برای دستیابی به آن باقی ماند، پروژه جدیدی که توسط موسسه ملی تحقیقات ژنوم انسانی در ایالات متحده توسعه یافته بود ظاهر شد. پروژه موسوم به ENCODE یک مجموعه داده مشترک از عناصر عملکردی ژنوم انسان است که از فناوریهای توالییابی DNA نسل بعدی و آرایههای کاشیکاری ژنومی استفاده میکند، فناوریهایی که میتوانند به طور خودکار مقادیر زیادی داده را با هزینه هر پایه بهطور چشمگیری تولید کنند. اما با همان دقت (خطای تماس پایه) و وفاداری (خطای اسمبلی).
پیشبینی عملکرد ژن
در حالی که حاشیه نویسی ژنوم اساساً بر اساس شباهت توالی (و در نتیجه همسانی ) است، از دیگر ویژگی های توالی ها می توان برای پیش بینی عملکرد ژن ها استفاده کرد. در واقع، بیشتر روشهای پیشبینی عملکرد ژن بر روی توالیهای پروتئین تمرکز میکنند، زیرا آموزندهتر و غنیتر از ویژگیها هستند. به عنوان مثال، توزیع اسیدهای آمینه آبگریز، بخش های گذرنده در پروتئین ها را پیش بینی می کند . با این حال، پیشبینی عملکرد پروتئین میتواند از اطلاعات خارجی مانند دادههای بیان ژن (یا پروتئین)، ساختار پروتئین یا برهمکنشهای پروتئین-پروتئین نیز استفاده کند . [26]
زیست شناسی تکاملی محاسباتی
اطلاعات بیشتر: فیلوژنتیک محاسباتی
زیست شناسی تکاملی مطالعه منشا و هبوط گونه ها و جزوه بیوانفورماتیک تغییر آنها در طول زمان است. انفورماتیک به زیست شناسان تکاملی کمک کرده است و محققان را قادر می سازد تا:
ردیابی تکامل تعداد زیادی از ارگانیسمها با اندازهگیری تغییرات در DNA آنها ، نه از طریق طبقهبندی فیزیکی یا مشاهدات فیزیولوژیکی به تنهایی،
مقایسه کل ژنوم ها ، که امکان مطالعه رویدادهای تکاملی پیچیده تر، مانند تکرار ژن ، انتقال افقی ژن ، و پیش بینی عوامل مهم در گونه زایی باکتریایی را فراهم می کند .
ساخت مدلهای ژنتیکی جمعیت محاسباتی پیچیده برای پیشبینی نتیجه سیستم در طول زمان [27]
ردیابی و به اشتراک گذاری اطلاعات در مورد تعداد فزاینده ای از گونه ها و موجودات
کار آینده تلاش می کند تا درخت زندگی را که اکنون پیچیده تر است بازسازی کند . [ به گفته چه کسی؟ ]
حوزه تحقیق در علوم کامپیوتر که از الگوریتمهای ژنتیک استفاده میکند ، گاهی با زیستشناسی تکاملی محاسباتی اشتباه گرفته میشود، اما این دو حوزه لزوماً به هم مرتبط نیستند.
ژنومیک مقایسه ای
مقاله اصلی: ژنومیک مقایسه ای
هسته اصلی تجزیه و تحلیل ژنوم مقایسه ای برقراری ارتباط بین ژن ها ( تحلیل ارتولوژی ) یا سایر ویژگی های ژنومی در موجودات مختلف است. این نقشه های بین ژنومی است که ردیابی فرآیندهای تکاملی مسئول واگرایی دو ژنوم را ممکن می کند. انبوهی از رویدادهای تکاملی که در سطوح مختلف سازمانی عمل میکنند، تکامل ژنوم را شکل میدهند. در پایین ترین سطح، جهش های نقطه ای بر نوکلئوتیدهای فردی تأثیر می گذارد. در سطح بالاتر، بخش های کروموزومی بزرگ دچار تکرار، انتقال جانبی، وارونگی، جابجایی، حذف و درج می شوند. [28] در نهایت، کل ژنوم ها در فرآیندهای هیبریداسیون، پلی پلوئیدی شدن و اندوسیمبیوز نقش دارند.، اغلب منجر به گونه زایی سریع می شود. پیچیدگی تکامل ژنوم، چالشهای هیجانانگیزی را برای توسعهدهندگان مدلها و الگوریتمهای ریاضی ایجاد میکند، که به طیفی از تکنیکهای الگوریتمی، آماری و ریاضی، از دقیق، اکتشافی ، پارامتر ثابت و الگوریتمهای تقریبی برای مدلهای مبتنی بر مدلهای مارکوف جزوه بیوانفورماتیک میشوند. الگوریتم های زنجیره ای مونت کارلو برای تحلیل بیزی مسائل بر اساس مدل های احتمالی
بسیاری از این مطالعات بر اساس تشخیص همسانی توالی برای اختصاص توالی به خانواده های پروتئینی است. [29]
پان ژنومیک
نوشتار اصلی: پان ژنوم
پان ژنومیک مفهومی است که در سال 2005 توسط تتلین و مدینی معرفی شد و سرانجام در بیوانفورماتیک ریشه دوانید. ژنوم پان مجموعه کامل ژنی یک گروه طبقه بندی خاص است: اگرچه در ابتدا برای سویه های نزدیک یک گونه به کار می رود، اما می توان آن را در زمینه بزرگتری مانند جنس، شاخه و غیره به کار برد. این ژنوم به دو بخش تقسیم می شود: ژنوم هسته: مجموعهای از ژنهای مشترک برای همه ژنومهای مورد مطالعه (اینها اغلب ژنهای خانهداری هستند که برای بقا حیاتی هستند) و ژنوم قابل انعطاف/منعطف: مجموعهای از ژنهایی که در همه ژنومها به جز یک یا برخی از ژنومهای مورد مطالعه وجود ندارند. یک ابزار بیوانفورماتیک BPGA می تواند برای مشخص کردن ژنوم پان گونه های باکتری استفاده شود. [30]
ژنتیک بیماری
مقاله اصلی: مطالعات ارتباط ژنومی
با ظهور توالی یابی نسل بعدی، ما در حال به دست آوردن داده های توالی کافی برای نقشه برداری از ژن های بیماری های پیچیده از جمله ناباروری ، [31] سرطان سینه [32] یا بیماری آلزایمر هستیم . [33] مطالعات ارتباط گسترده ژنوم یک رویکرد مفید برای مشخص کردن جهشهای مسئول چنین بیماریهای پیچیدهای است. [34] از طریق این مطالعات، هزاران نوع DNA شناسایی شده اند که با بیماری ها و ویژگی های مشابه مرتبط هستند. [35]علاوه بر این، امکان استفاده از ژن ها در پیش آگهی، تشخیص یا درمان یکی از ضروری ترین کاربردها است. بسیاری از مطالعات در مورد راههای امیدوارکننده برای انتخاب ژنهای مورد استفاده و مشکلات و مشکلات استفاده از ژنها برای پیشبینی حضور یا پیشآگهی بیماری بحث میکنند. [36]
مطالعات ارتباط ژنومی با موفقیت هزاران گونه ژنتیکی رایج را برای بیماری ها و صفات پیچیده شناسایی کرده است. با این حال، این گونه های رایج تنها بخش کوچکی از وراثت پذیری را توضیح می دهند. [37] گونههای نادر ممکن است برخی از وراثتپذیریهای گمشده را توجیه کنند. [38] مطالعات توالی یابی کل ژنوم در مقیاس بزرگ به سرعت میلیون ها ژنوم کامل را توالی یابی کرده اند و چنین مطالعاتی صدها میلیون گونه نادر را شناسایی کرده است. [39] حاشیه نویسی عملکردیپیشبینی اثر یا عملکرد یک نوع ژنتیکی و کمک به اولویتبندی انواع عملکردی نادر، و گنجاندن این حاشیهنویسیها میتواند به طور موثر قدرت ارتباط ژنتیکی تجزیه و تحلیل واریانتهای نادر را در مطالعات توالییابی کل ژنوم افزایش دهد. [40] برخی از ابزارها برای ارائه تجزیه و تحلیل تداعی انواع نادر یکپارچه برای داده های توالی یابی کل ژنوم، از جمله ادغام داده های ژنوتیپ و حاشیه نویسی های عملکردی آنها، تجزیه و تحلیل ارتباط، خلاصه نتایج و تجسم ایجاد شده اند. [41] [42]
تجزیه و تحلیل جهش در سرطان
مقاله اصلی: انکوژنومیکس
در سرطان ، ژنوم سلولهای آسیبدیده به روشهای پیچیده یا حتی غیرقابل پیشبینی بازآرایی میشوند. تلاشهای توالییابی گسترده برای شناسایی جهشهای نقطهای ناشناخته قبلی در انواع ژنهای سرطان استفاده میشود. بیوانفورماتیکان به تولید سیستمهای خودکار تخصصی برای مدیریت حجم عظیم دادههای توالی تولید شده ادامه میدهند و الگوریتمها و نرمافزارهای جدیدی را برای مقایسه نتایج توالییابی با مجموعه رو به رشد توالیهای ژنوم انسانی و پلیمورفیسمهای مولفه ایجاد میکنند. فنآوریهای جدید تشخیص فیزیکی، مانند ریزآرایههای الیگونوکلئوتیدی برای شناسایی سود و زیان کروموزومی (به نام هیبریداسیون مقایسهای ژنومی ) استفاده میشوند.، و آرایه های پلی مورفیسم تک نوکلئوتیدی برای تشخیص جهش های نقطه ای شناخته شده . این روشهای تشخیص همزمان چند صد هزار مکان را در جزوه بیوانفورماتیک ژنوم اندازهگیری میکنند، و هنگامی که در توان عملیاتی بالا برای اندازهگیری هزاران نمونه استفاده میشوند، در هر آزمایش ، ترابایت داده تولید میکنند. باز هم مقادیر انبوه و انواع جدید داده ها فرصت های جدیدی را برای بیوانفورماتیکان ایجاد می کند. دادهها اغلب حاوی تغییرات یا نویز قابلتوجهی هستند و بنابراین مدل Hidden Markov و روشهای تحلیل نقطه تغییر برای استنتاج تغییرات تعداد کپی واقعی توسعه مییابند .
دو اصل مهم را می توان در تجزیه و تحلیل ژنوم سرطان از نظر بیوانفورماتیک مربوط به شناسایی جهش در اگزوم استفاده کرد. اول، سرطان بیماری جهشهای جسمی انباشته شده در ژنها است. سرطان دوم حاوی جهش های راننده است که باید از [43]
[44]
()، ()، ()، -“” () /[45] :
فهرست مطالب